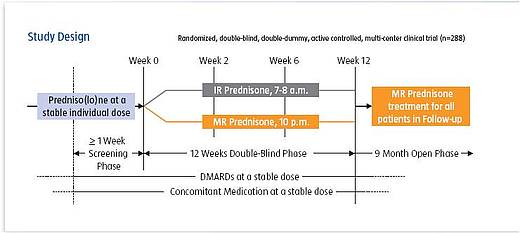
CAPRA-1 (Circadian Administration of Prednisone in Rheumatoid Arthritis) ist eine 12-wöchige, zulassungsrelevante, randomisierte, durch eine aktive Vergleichssubstanz kontrollierte internationale multizentrische Phase-III-Studie mit einer anschließenden offenen Phase über 9 Monate zur Prüfung der Wirksamkeit und Verträglichkeit einer neuen Prednison-Formulierung (Prednison MR, Lodotra) in einer Dosierung von 3-10 mg/Tag im Vergleich zu herkömmlichem Prednison bei Patienten mit einer rheumatoiden Arthritis (RA).
Studienziele waren die Verringerung der Morgensteifigkeit, die Reduktion von Interleukin-6 (IL-6) im Serum und die Verbesserung der klinischen Symptome einer aktiven RA, außerdem die Frage der Sicherheit und Verträglichkeit der neuen Substanz.
Als primärer Endpunkt wurde die relative Veränderung der Dauer der Morgensteifigkeit nach 12 Wochen im Vergleich zum Ausgangsbefund definiert.
Sekundäre Endpunkte waren
- die Verringerung von Interleukin-6 (IL-6) im Serum nach 12 und 52 Wochen
- die Verbesserung im DAS-28 nach 12 und 52 Wochen
- die ACR-20-Ansprechrate nach 12 und 52 Wochen
- die Einzelkomponenten des ACR-20 nach 12 und 52 Wochen
- die Verringerung der Schmerzintensität über den Tag
- die Verbesserung der Schlafqualität
- die Verbesserung der funktionellen Kapazität (HAQ) und der Lebensqualität (SF-36)
- Veränderungen von Osteocalcin im Serum sowie
- die Sicherheit und Verträglichkeit von Prednison MR.
Aufgenommen in die Studie wurden 288 erwachsene RA-Patienten beiderlei Geschlechts im Alter von 18 bis 80 Jahren (Klassifikation der RA nach den ACR-Kriterien von 1987). Alle Patienten mussten eine aktive Erkrankung aufweisen, d.h. eine Morgensteifigkeit von mindestens 45 Minuten, Schmerzen von mindestens 30 mm auf einer visuellen Analogskala (VAS) von 1-100, mindestens 3 druckschmerzhafte Gelenke, mindestens 1 geschwollenes Gelenk sowie eine BSG (nach Westergren) von ≥ 28 mm/h oder ein CRP von mehr als dem 1,5-fachen des Normbereichs.
Die Patienten hatten vor Einschluß in die Studie Glucocorticoide (Cortisonpräparate) über mindestens drei Monate erhalten, davon mindestens einen Monat vor der Randomisierung in einer konstanten Dosis zwischen 2,5 und 10 mg Prednisonäquivalent. Ebenso waren sie zuvor über mindestens 3 Monate mit einer krankheitsmodifizierenden Substanz (DMARD, langwirksame antirheumatische Therapie, früher sogenannte Basistherapie) behandelt worden, auch dabei einen Monat lang vor der Randomisierung in konstanter Dosis. Bei Unverträglichkeit von DMARDs war auch eine Teilnahme ohne ein solches Präparat möglich. Biologika waren in den 4 Monaten vor und während der Studie nicht erlaubt.
Die Behandlung mit krankheitsmodifizierenden Substanzen und mit nicht-steroidalen Antiphlogistika (NSAIDs; non steroidal anti inflammatory drugs, cortisonfreie Entzündungshemmer) wurde während der Doppelblindphase unverändert weitergeführt. Glucocorticoide (außer der Prüfsubstanz), intraartikuläre Injektionen, Synoviorthesen und Kryotherapie waren nicht zulässig.
Die Patienten wurden in einem Verhältnis von 1:1 auf die zwei Studienarme aufgeteilt („randomisiert“):
- 144 Patienten erhielten Prednison MR (modified release, verzögerte Freisetzung) in einer stabilen Dosis von 3-10 mg pro Tag entsprechend dem Prednison-Äquivalent vor Eintritt in die Studie
- 144 Patienten erhielten herkömmliches Prednison (Prednison IR, immediate release, sofortige Freisetzung) in einer stabilen Dosis von 3-10 mg pro Tag entsprechend dem Prednisondosis-Äquivalent vor Eintritt in die Studie
Der einzige Unterschied zwischen den beiden Behandlungsarmen war nach dem gewählten Design der Zeitpunkt, zu dem das Prednison freigesetzt wurde: Etwa gegen 2:00 Uhr morgens bei der MR-Formulierung (Einnahmezeitpunkt ca. 22:00 Uhr) und zwischen 6:00 und 8:00 Uhr bei der konventionellen Prednison-Gruppe (Einnahmezeitpunkt zwischen 6:00 und 8:00 Uhr). Die MR-Gruppe erhielt zusätzlich morgens, die IR-Gruppe abends Placebotabletten.
Nach Abschluss der doppelblinden Phase hatten alle Patienten die Möglichkeit, an einer 9-monatigen offenen Phase mit Prednison MR in identischer Dosis zur Vortherapie teilzunehmen.
Zu Studienbeginn wiesen die Patienten mit durchschnittlich etwa 55 Jahren das typische Alter auf, das man in vergleichbaren Studienpopulationen sieht. Im Mittel lag die Krankheitsdauer bei 9,6 Jahren, d.h. die Erkrankung war zu diesem Zeitpunkt bereits fortgeschritten. Mehr als 94% wurden bei Einschluß in die Studie mit DMARDs behandelt. 80% erhielten cortisonfreie Entzündungshemmer (NSAIDs, nicht-steroidale Antirheumatika).
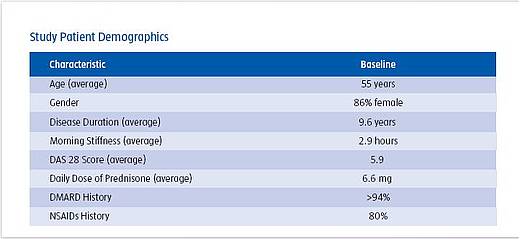
Die Erkrankung war vor Beginn der Studie mit einer Morgensteifigkeit von durchschnittlich 2,9 Stunden und einem DAS-28 von 5,9 hochaktiv.
Die mittlere Prednisondosis lag initial bei 6,6 mg/Tag.
Nach 12 Wochen konnten Baseline- und Endpunkt-Daten für die Morgensteifigkeit bei 125 Patienten der MR-Prednison-Gruppe (87%) und bei 129 Patienten (90%) der IR-Prednison-Gruppe ausgewertet werden.
Nach diesem Behandlungszeitraum von 12 Wochen verringerte sich die Morgensteifigkeit unter Prednison MR um 22,7% (44 Minuten) gegenüber 0,4% unter herkömmlichem Prednison (p < 0,05). Der absolute Unterschied zwischen MR- und IR-Gruppe von 29 Minuten war nicht nur statistisch signifikant, sondern auch klinisch relevant.
Nach Wechsel auf Prednison MR in der offenen Verlängerungsstudie kam es auch in der Gruppe der Patienten, die ursprünglich konventionelles Prednison erhalten hatten, zu einer signifikanten Verkürzung der Morgensteifigkeit.
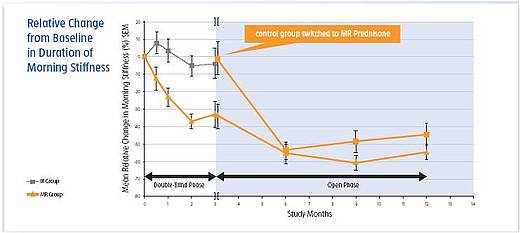
Die Wirkung der neuen Prednison-MR-Formulierung trat sehr schnell ein. Ein signifikanter Unterschied in der Dauer der Morgensteifigkeit zeigte sich bereits nach zwei Wochen mit einem Unterschied von 10% zwischen den beiden Therapiearmen mit einem Vorteil für die MR-Gruppe. Mit zunehmender Behandlungsdauer verstärkte sich dieser Unterschied und erreichte nach 7 Wochen bis zum Ende der verblindeten Phase nach 12 Wochen ein Plateau.
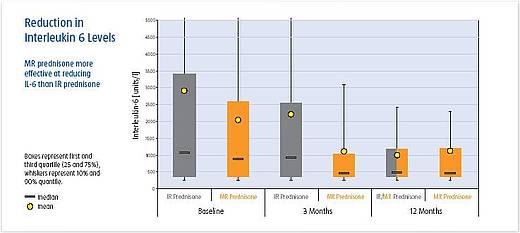
Parallel zur Verringerung der Morgensteifigkeit kam es unter Prednison MR auch zu einem deutlichen Absinken des Interleukin-6-Spiegels im Serum. Nach 12 Wochen war IL-6 im Vergleich zum Ausgangspunkt in der MR-Gruppe um 160 IU/l abgefallen (29%), während sich in der traditionell behandelten Gruppe im Verlauf über 12 Wochen keine Veränderung des IL-6-Spiegels im Serum zeigte. Die Unterschiede zwischen Prednison MR und Prednison IR waren signifikant (p< 0.05).
Dieser Effekt setzte sich über die 9-monatige offene Verlängerungsphase fort.
Bei allen übrigen sekundären Endpunkten zeigten sich zwischen den beiden Präparaten keine signifikanten Unterschiede.
Die Verträglichkeit und Sicherheit von Prednison MR entsprach in der CAPRA-1-Studie den Daten, die von der Therapie mit Glukocorticoiden bekannt sind.
Insgesamt wurden bei 13% der Prednison-MR-Patienten behandlungsbedingte unerwünschte Ereignisse dokumentiert gegenüber 11% unter Prednison IR. Ein Therapieabbruch wegen unerwünschten Ereignissen war in 8% unter Prednison MR und in 7% unter Prednison IR notwendig. Schwere unerwünschte Ereignisse traten selten auf (3% unter Prednison MR, 2% unter Prednison IR). Signifikante Unterschiede zwischen der neuen Substanz und dem herkömmlichen Cortison bestanden nicht.
Häufigste unverwünschte Ereignisse waren Kopfschmerzen (4% unter Prednison MR, 3% unter Prednison IR), Schmerzen im Oberbauch (4% bzw. 6%), Übelkeit (4% bzw. 3%), banale Infekte der oberen Luftwege (Nasopharyngitis, 3% bzw. 6%), flüchtige Hautrötung („Flush“, 3% bzw. 4%), Schmerzen im Brustbereich (2% in beiden Gruppen), Blutdruckerhöhung (Hypertonie, 2% in beiden Gruppen), Schwindel (1% bzw. 3%), Verdauungsbeschwerden (Dyspepsie, 1% bzw. 2%) sowie Bronchitis (1% bzw. 4%). Dabei zeigten sich ebenfalls keine signifikanten Unterschiede zwischen der neuen Cortisonformulierung und dem traditionellen Prednison.
Schlussfolgerung
Die Autoren der CAPRA-1-Studie kommen zu dem Ergebnis, daß die neue MR-Prednison-Formulierung der traditionellen Verabreichungsform bei der Reduktion der Morgensteifigkeit und bei der Reduktion der Interleukin-6-Spiegel im Serum überlegen ist. Dabei führte die abendliche Verabreichung der Cortisondosis in der neuen Form nicht zu einer Erhöhung der Nebenwirkungsrate gegenüber der morgendlichen Einnahme.
Mit Prednison MR kann nach dieser Studie erstmals der Wirkstoff Prednison zum physiologisch optimalen Zeitpunkt freigesetzt werden. Die daraus resultierende effektive Verkürzung der Morgensteifigkeit verbessere insbesondere morgens die funktionelle Kapazität und trage damit zu einer erheblichen Verbesserung der Lebensqualität bei.
Prednison stelle in der MR-Formulierung einen wesentlichen Fortschritt bei der Behandlung der rheumatoiden Arthritis gegenüber herkömmlichen Glucocorticoiden dar. Durch die Umsetzung der chronobiologischen Erkenntnisse zum Pathomechanismus der RA in eine zeitoptimierte Prednisongabe trage Prednison MR zu einer wesentlichen Optimierung der Cortisontherapie bei.
Die Autoren schließen mit dem Ausblick, daß über den Einsatz bei der rheumatoiden Arthritis hinaus die neue Tablette auch für die Behandlung anderer Krankheiten wie z.B. Polymyalgia rheumatica oder für die Asthmatherapie von Bedeutung sein kann.
Referenzen:
Buttgereit F, Doering G, Schaeffler A, Witte St, Sierakowski St, Gromnica-Ihle E, Jeka S, Krueger K, Szechinski J, Alten R:
Efficacy of modified-release versus standard prednisone to reduce duration of morning stiffness of the joints in rheumatoid arthritis (CAPRA-1): a double-blind, randomised controlled trial
The Lancet, Volume 371, Issue 9608, Pages 205 - 214, 19 January 2008
Abstract:
Volltext: Kein open access zum Volltext
Buttgereit F, Doering G, Knauer Chr, Witte St, Szechinski J, Alten,R:
Prednisone Chronotherapy of Rheumatoid Arthritis: Sustained Efficacy of a Novel Modified-Release Formulation Over 12 Months
ACR Philadelphia 2009, Abstr. 409
Abstract: