TOWARD (Tocilizumab in cOmbination With traditional DMARD therapy) ist eine randomisierte, doppelblinde, placebo-kontrollierte internationale Multicenter-Phase III-Studie zur Prüfung der Wirksamkeit und Sicherheit von Tocilizumab in einer Dosierung von 8 mg/kg in Kombination mit herkömmlichen krankheitsmodifizierenden Therapien (DMARDs) bei 1.216 Erwachsenen mit einer aktiven rheumatoiden Arthritis und unzureichendem Ansprechen auf herkömmliche DMARDs (Methotrexat (MTX), Chloroquin, Hydroxychloroquin, parenterales Gold, Sulfasalazin, Azathioprin oder Leflunomid). Die Studie wurde an 130 Studienzentren in 18 Ländern einschließlich der USA durchgeführt [1].
Zusammenfassung der Ergebnisse
Die ACR-Ansprechraten in der TOWARDStudie waren unter Tocilizumab unabhängig von der Wahl der Hintergrundtherapie mit DMARDs höher als unter Placebo plus DMARD
Tocilizumab verminderte in Kombination mit einer DMARD-Therapie die Symptome der RA und führte bei signifikant mehr Patienten zu einer Remission als in der Kontrollgruppe.
Zudem erfuhren Patienten der Tocilizumab-Gruppe auch Verbesserungen der körperlichen Funktionsfähigkeit und der Lebensqualität, was für die Tocilizumab plus DMARD Kombinationstherapie im Vergleich zu einer alleinigen DMARD-Therapie spricht.
Die Studie im Detail
Studienziel
Wirksamkeit und Sicherheit von Tocilizumab in einer Dosierung von 8 mg/kg in Kombination mit herkömmlichen krankheitsmodifizierenden Therapien (DMARDs)
Einschlußkriterien
Mittelschwere bis schwere RA von mindestens 6 Monaten Dauer
Unzureichendes Ansprechen auf herkömmliche DMARD-Therapien
Ausschlußkriterien
Erfolglose Vorbehandlung mit TNF-alpha-Inhibitoren
Studiendesign
Zweiarmige, 1:2 randomisierte Studie über 24 Wochen:
Tocilizumab 8 mg/kg KG alle 4 Wochen plus DMARDs (n=803)
Placebo plus DMARDs (n=413)
Rescue-Therapie (Anpassung der Hintergrund-DMARD-Dosis und/oder Behandlung mit einem anderen konventionellen DMARD) bei Patienten, bei denen nach 16 Wochen im Gelenkstatus (Zahl geschwollener und Zahl schmerzhafter Gelenke) keine Verbesserung von mindestens 20 % vom Ausgangswert erzielt wurde.
Studienergebnisse
Studienpopulation
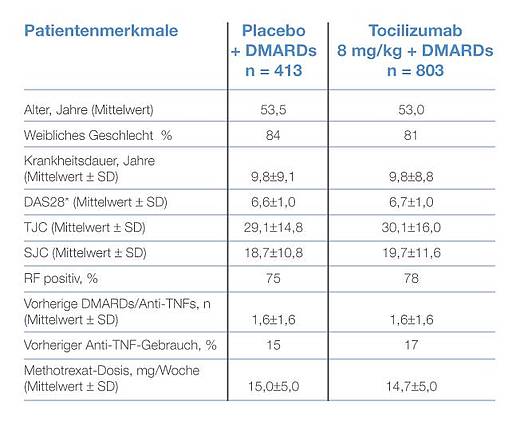
Die demographischen und krankheitsbezogenen Charakteristika zum Ausgangszeitpunkt waren zwischen der Tocilizumab- und der MTX-Gruppe vergleichbar (Tab. 1). Die Patienten waren bei Aufnahme in die Studie im Mittel 53 Jahre alt und 9,8 Jahre an RA erkrankt.
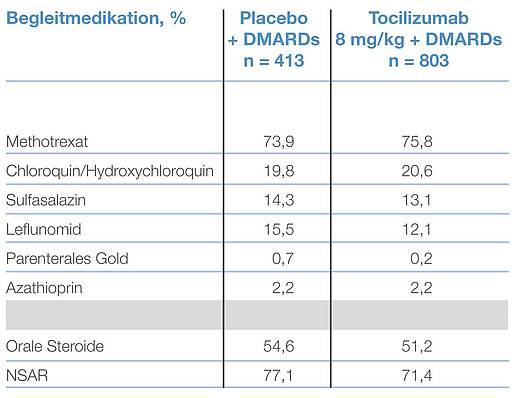
Die medikamentöse DMARD-Begleittherapie bestand zu 73,9 bzw. 75,8 Prozent aus MTX. Zweithäufigstes DMARD war Chloroquin/Hydroxychloroquin bei 19,8 bzw. 20,6 Prozent. Orale Steroide wurden bei rund der Hälfte der Studienpatienten eingesetzt (Tab. 2). 76 % wurden mit einem DMARD, 23 % mit zwei oder mehr DMARD behandelt.
Ansprechraten
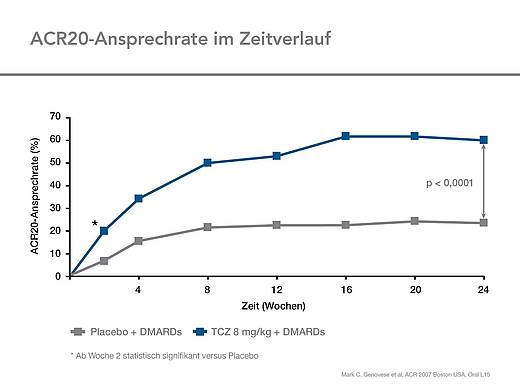
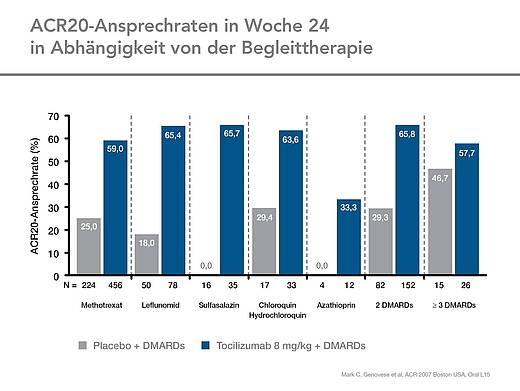
Im Vergleich zu Placebo erreichten signifikant mehr Patienten in der Tocilizumab-Gruppe ein ACR20-Ansprechen nach 24 Wochen (Abb. 1).
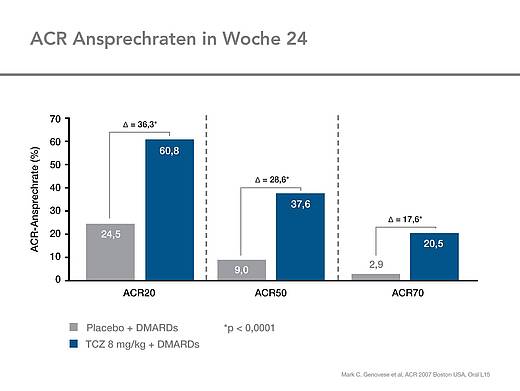
Im Einzelnen betrugen die Verbesserungsraten für den ACR 20: 60,8 % vs 24,5 %, für den ACR50: 37,6 vs 9,0 % und den ACR70: 20,5 vs 2,9 % (Abb.2).
Das Ansprechen war unabhängig von der Art und Menge der zuvor nicht ausreichend wirksamen DMARDs [1, 2].
Unter Tocilizumab konnte in Kombination mit jedem der in der Studie zugelassenen DMARDs eine höhere ACR20-Ansprechrate erzielt werden als mit Placebo plus DMARDs [1, 2] (Abb. 3). Unterschiede in den Ansprechraten waren selbst dann erkennbar, wenn zwei DMARDs als Hintergrundtherapie eingesetzt wurden, was in einer Subgruppe von 234 Patienten der Fall war. Hier betrug die ACR20-Ansprechrate unter Tocilizumab 66 % im Gegensatz zu 29 % unter Placebo [1, 2].
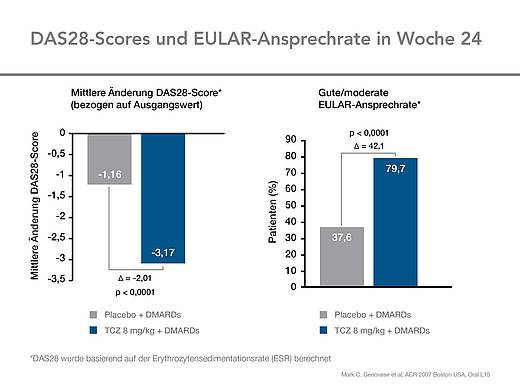
Die deutliche klinische Verbesserung unter der Behandlung mit Tocilizumab plus DMARD(s) im Vergleich mit Placebo plus DMARD(s) führte konsequent auch zu einer ausgeprägten Verbesserung im DAS28 (Abb. 4). Die mittleren Veränderungen im DAS28 vom Basiswert aus lagen unter Tocilizumab bei -3,17 verglichen mit -1,16 unter Placebo. In Woche 24 erreichten unter Tocilizumab signifikant mehr Patienten ein gutes und moderates Ansprechen gemäß den EULAR-Kriterien (Abb. 4).
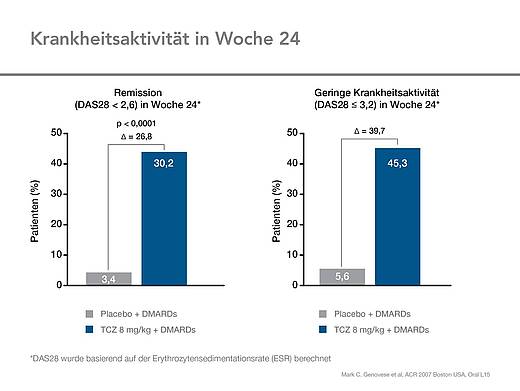
Remissionsraten
Knapp ein Drittel der Patienten unter Tocilizumab (30,2 %) erreichte nach 24 Wochen Behandlung eine DAS28-Remission (DAS28 < 2,6), in der Placebo plus DMARDs-Gruppe waren es 3,4 %. Eine geringe Krankheitsaktivität (DAS ≤ 3,2) wiesen unter Tocilizumab 54,3 Prozent der Patienten auf, unter Placebo plus DMARDs waren es 5,6 Prozent (Abb. 5).
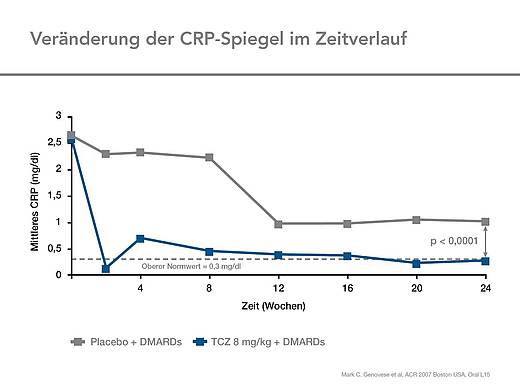
Entzündungsparameter und Hämoglobinspiegel
Die mittlere Reduktion des CRP war nach 24 Wochen signifikant größer als unter Placebo (-2.20 vs -0.27 mg/dl) (Abb. 6).
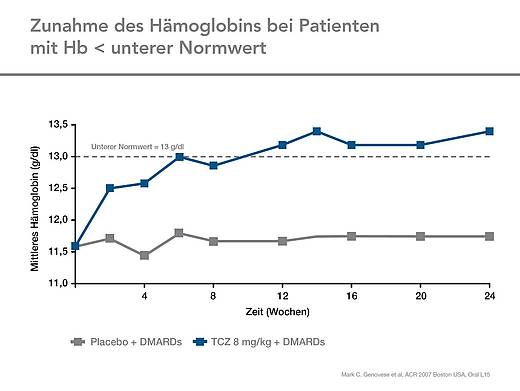
Die Hämoglobinspiegel waren unter Tocilizumab nach 24 Wochen um 0,98 g/dl angestiegen verglichen mit -0.13 g/dl in der Placebogruppe (Abb. 7). Der Hämoglobinwert stieg unter Tocilizumab schnell an und normalisierten sich bei den Patienten, die zu Studienbeginn anämisch gewesen waren.
Patient Reported Outcomes (PROs)
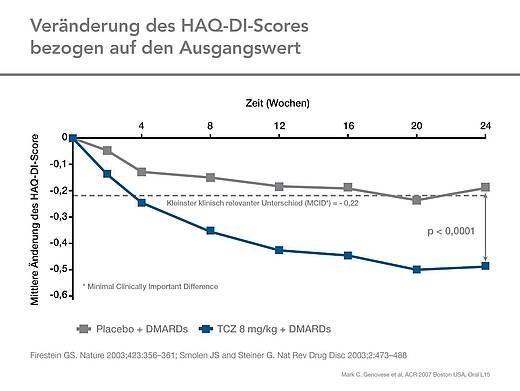
In der Tocilizumab-Gruppe wurde bei 60 Prozent im Vergleich zu 34 Prozent der Placebo-Patienten eine klinisch relevante Verbesserung der HAQ-DI (Abb. 8), der FACIT-Fatigue und der Short-Form-36 (SF-36) Scores dokumentiert [1, 2].
Sicherheit und Verträglichkeit
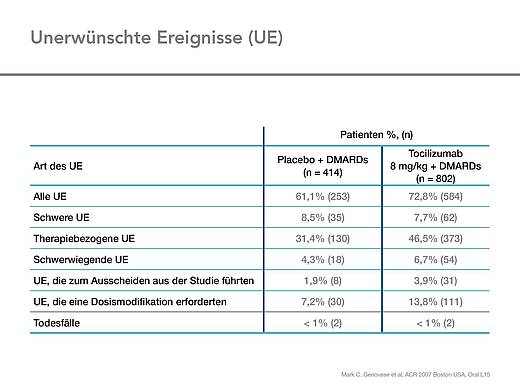
Die Größe der Studienpopulation erlaubte das Aufdecken wesentlicher Sicherheitsbelange für die Kombination von Tocilizumab mit den DMARDs Methotrexat, Leflunomid, Antimalariamedikamenten und Sulfasalazin. Trotz der verschiedenen DMARDs als Kombinationspartner stimmten Häufigkeit und Schweregrad der unerwünschten Wirkungen (UE) mit vorangegangenen Berichten überein [1].
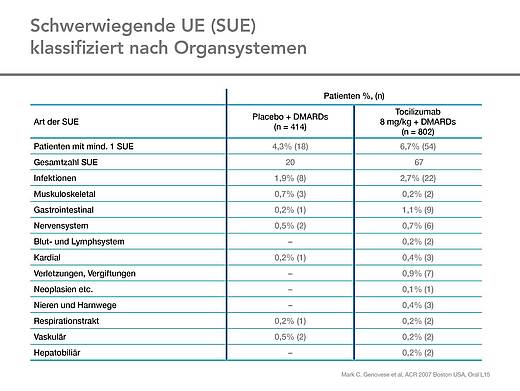
Die häufigste UE waren Infektionen des oberen Respirationstraktes. Die Gesamtinzidenz schwerer unerwünschter Wirkungen (SUE) war niedrig und nur wenige Patienten mit schweren Infekten mussten die Studie aus diesem Grund abbrechen (Tab. 3 und 4) [1].
Zu den UEs, die in der Tocilizumab-Gruppe häufiger auftraten, zählten gastrointestinale Beschwerden und Hautausschläge. Bei den GI-Komplikationen war auch immer ein Risikofaktor, wie die Behandlung mit NSAR oder Glukokortikoiden vorhanden. Die Hautausschläge wurden nicht als Hypersensitivitätsreaktionen gesehen.
Insgesamt traten in der Tocilizumab-Gruppe mehr unerwünschte Wirkungen (UE) auf als in der Kontrollgruppe (Tab. 3). Mehr als 90 Prozent der UEs in den beiden Gruppen waren leichter oder mittelschwerer Natur. Schwerwiegende unerwünschte Wirkungen und UEs, die zum Therapieabbruch bzw. zur Dosismodifikation führten, waren in der Tocilizumab-Gruppe häufiger als in der Kontrollgruppe (Tab. 3 und 4). Die meisten Patienten mit unerwünschten Ereignissen erhielten Methotrexat als Begleit-DMARD, gefolgt von Patienten, die zusätzlich mit einer DMARD-Kombination behandelt wurden und der Population, die mit Leflunomid als Begleit-DMARD behandelt wurde. Ein kausaler Zusammenhang kann allerdings nicht gesehen werden, da diese Patientengruppen jeweils auch am größten waren [1].
Literatur
Genovese MC, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: The Tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study.
Arthritis Rheum 2008; 58:2968–2980 LinkACR 2007 – L15 Genovese M C et al., IL-6 receptor inhibition with tocilizumab reduces disease activity in patients with rheumatoid arthritis with inadequate response to arrange of DMARDs: the TOWARD study, ACR 2007, Abstract L15
Fachinformation RoActemra®. Stand Juli 2011